University of California, Berkeley scientists have for the first time used CRISPR-Cas9 gene editing to disable a defective gene that causes amyotrophic lateral sclerosis, or Lou Gehrig’s disease, in mice, extending their lifespan by 25 percent.
Credit: David Schaffer
The therapy delayed the onset of the muscle wasting that characterizes the disease, which results in progressive weakness and eventually proves fatal when the muscles that control breathing fail.
This step toward a CRISPR cure for human ALS will be reported Dec. 20 in the journal Science Advances.
The mice were genetically engineered to express a mutated human gene that in humans causes about 20 percent of all inherited forms of the disease and about 2 percent of all cases of ALS worldwide. Though the genetic cause is not known for all cases of ALS, all are accompanied by the premature death of motor neurons in the brain stem and spinal cord. The neurons allow the brain to control muscles, so loss of this connection means loss of muscle control.
“Being able to rescue motor neurons and motor neuron control over muscle function, especially the diaphragm, is critically important to being able to not only save patients, but also maintain their quality of life,” said senior author David Schaffer, a professor of chemical and biomolecular engineering and director of the Berkeley Stem Cell Center.
The devastating disease usually strikes people between the ages of 40 and 70. An estimated 20,000 Americans are afflicted, and there are no treatments to slow the muscle degeneration.
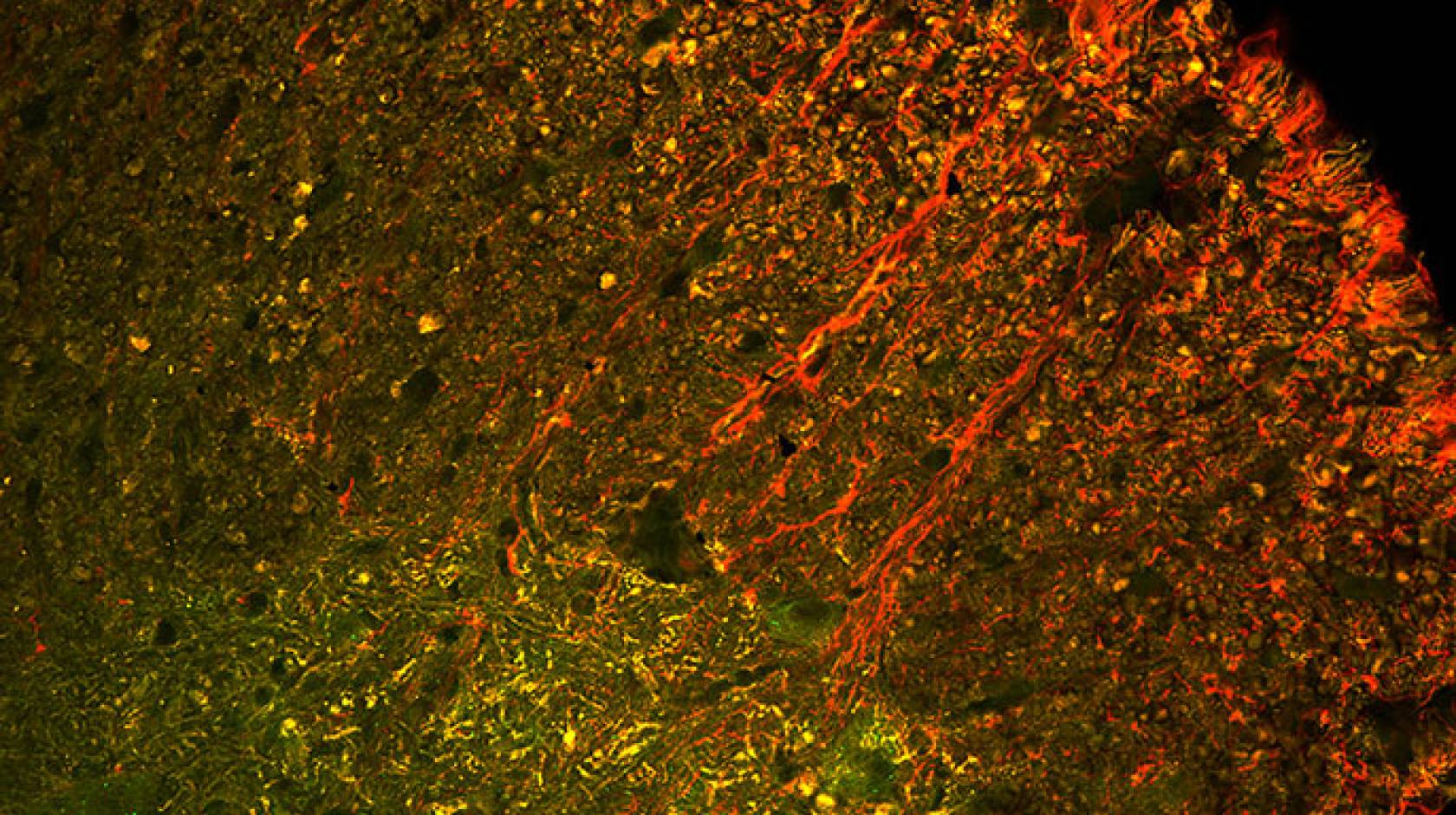
The UC Berkeley research team used a virus that Schaffer’s team engineered to seek out only motor neurons in the spinal cord and deliver a gene encoding the Cas9 protein into the nucleus. There, the gene was translated into the Cas9 protein, a molecular scissors that cut and disabled the mutant gene responsible for ALS.
Treatment delays onset of symptoms, extends life
In this case, Cas9 was programmed to knock out only the mutated gene SOD1 (superoxide dismutase 1). The onset or start of the disease was delayed by almost five weeks, and mice treated by the gene therapy lived about a month longer than the typical four-month lifespan of mice with ALS. Healthy mice can live a couple of years.
The researchers found that, at death, the only surviving motor neuron cells in the mice were those that had been “infected” with the virus and contained Cas9 protein, said Thomas Gaj, a postdoctoral fellow who led the study, now at the University of Illinois at Urbana-Champaign.
“The treatment did not make the ALS mice normal and it is not yet a cure,” Schaffer cautioned. “But based upon what I think is a really strong proof of concept, CRISPR-Cas9 could be a therapeutic molecule for ALS. When we do additional optimization of the delivery to get CRISPR-Cas9 into an even higher percentage of cells, we think we are going to see even better increases in lifespan.”
One of several challenges is to eliminate the SOD1 mutation in other brain and spinal cord cells that support motor neurons. Schaffer’s team is designing a version of the virus – a highly modified adeno-associated virus, or AAV – that will deliver the Cas9 gene to two types of glial cells, astrocytes and oligodendrocytes, that appear to take out neighboring motor neurons, effectively a “bystander effect.”
“I tend to be really cautious, but in this case I would be quite optimistic that if we are able to eliminate SOD1 within not just the neurons but also the astrocytes and supporting glia, I think we are going to see really long extensions of lifespan,” he said.
Schaffer also is working on a self-destruct switch for the Cas9 protein, so that once it knocks out the SOD1 gene, the Cas9 can be eliminated from the cell so as not to accidentally modify other genes or trigger an immune reaction.
Special delivery by adeno-associated virus
Schaffer has been working with the AAV virus for nearly 20 years, evolving it to target specific cells, like motor neurons, without infecting other types of cells. AAV is found in many if not all humans and primates, and appears to be benign.
“We have engineered new AAV vehicles that are capable of high-efficiency delivery to a number of cell and tissue targets in the body, and when CRISPR-Cas9 came along, we viewed it as a wonderful opportunity to put together this incredibly powerful cargo with the ability to carry that cargo to a number of cells and disease targets in vivo,” he said.
A modified AAV is likely to be approved soon by the Food and Drug Administration as a delivery vehicle for a gene therapy against a rare disease called Leber congenital amaurosis type 2, and other therapies delivered by AAV are in the pipeline, Schaffer said.
These therapies are currently based on natural versions of AAV, however, which are not optimized for high-efficiency delivery to most therapeutically important cell targets, so clinicians must either use massive doses or apply the AAV using an invasive surgery. Schaffer has developed technology to engineer viruses for targeted delivery to many cells and tissues following simple routes of administration, for example directly into tissue such as the eye and brain. This led him to cofound a company in 2014, 4D Molecular Therapeutics, to optimize AAV to target any tissue and carry a variety of gene therapies into cells.
“Researchers in the field really know we need better vectors that can target cells through a simple, logical route of administration, and can do so in a very, very efficient way,” he said. “Ours do.”
The research was supported by the National Institutes of Health. UC Berkeley co-authors of the paper are Thomas Gaj, David Ojala, Freja Ekman, Leah Byrne and Prajit Limsirichai.

