
An international team of researchers — led by scientists at UC San Francisco, Stanford University, the University of North Carolina, and the Friedrich-Alexander University Erlangen-Nürnberg in Germany — has developed a new opioid drug candidate that blocks pain without triggering the dangerous side effects of current prescription painkillers.
Their secret? Starting from scratch — with computational techniques that let them explore more than four trillion different chemical interactions.
In a new study — published online Aug. 17, 2016, in Nature — the researchers used the newly deciphered atomic structure of the brain’s “morphine receptor” to custom engineer a novel drug candidate that blocked pain as effectively as morphine in mouse experiments, but did not share the potentially deadly side effects typical of opioid drugs. In particular, the new drug did not interfere with breathing — the main cause of death in overdoses of prescription painkillers as well as street narcotics like heroin — or cause constipation, another common opioid side effect. The new drug also appears to sidestep the brain’s dopamine-driven addiction circuitry and did not cause drug-seeking behavior in mice.
More work is needed to establish that the newly formulated compound is truly non-addictive and to confirm that it is as safe and effective in humans as it is in rodents, the authors say. But if the findings are borne out, they could transform the fight against the ongoing epidemic of prescription painkiller addiction.
Deaths from opioid drug overdoses have been on the rise in the U.S. for decades. According to the Centers for Disease Control and Prevention, 28,000 Americans died of narcotic overdoses in 2014, four times more than in 1999, with more than half of these deaths involving prescription drugs. The epidemic has gotten the attention of national leaders: in February, 2016, President Obama proposed $1.1 billion in new funding for opioid addiction treatment, and in July Congress passed the Comprehensive Addiction and Recovery Act, a bill intended to curb opioid abuse and improve treatment.
But as damaging as opioids can be, modern medicine depends on these drugs as our most powerful weapon against pain.
“Morphine transformed medicine,” said Brian Shoichet, Ph.D., a professor of pharmaceutical chemistry in UCSF’s School of Pharmacy and co-senior author on the new paper. “There are so many medical procedures we can do now because we know we can control the pain afterwards. But it’s obviously dangerous too. People have been searching for a safer replacement for standard opioids for decades.”
Virtual experiments lead to novel opioid chemistry
Much of drug discovery, Shoichet says, begins by taking a successful drug like morphine and tweaking its structure to try to get rid of side effects while maintaining its primary function. The new study took a different, much more radical approach: “We didn’t want to just optimize chemistry that already existed,” Shoichet said. “We wanted to get new chemistry that would confer completely new biology.”
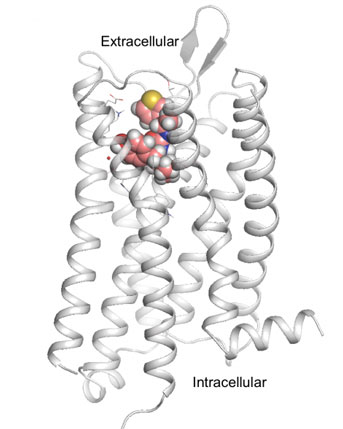
Credit: Anat Levit/UCSF
Key to the new paper was knowing the atomic structure of the mu-opioid receptor, the brain’s “morphine receptor,” which was recently deciphered by co-senior author and 2012 Nobel laureate Brian Kobilka, M.D., a professor of molecular and cellular physiology at the Stanford University School of Medicine.
“With traditional forms of drug discovery, you’re locked into a little chemical box,” Shoichet said. “But when you start with the structure of the receptor you want to target, you can throw all those constraints away. You’re empowered to imagine all sorts of things that you couldn’t even think about before.”
With this structural information in hand, Shoichet’s team turned to a computational approach called molecular docking, which was pioneered in the 1980s at UCSF’s School of Pharmacy by Shoichet’s mentor, emeritus professor Tack Kuntz, Ph.D. In a two-week period, the researchers performed roughly four trillion “virtual experiments” on a UCSF computer cluster, simulating how millions of different candidate drugs could turn and twist in millions of different angles to find those configurations that were most likely to fit into a pocket on the receptor and activate it. They also strove to avoid molecules that could stimulate beta-arrestin2, part of a biological pathway linked to the respiratory suppression and constipation typical of other opioids.
This led to a short-list of 23 candidate molecules judged by the software and the research team — especially co-lead authors Henry Lin, Ph.D. of UCSF and Aashish Manglik, M.D., Ph.D. at Stanford — to be most likely to activate the mu-opioid receptor in the way the researchers wanted.
Only then did the team actually test these candidate drugs in the real world. Co-lead author Dipendra Aryal, Ph.D., led a team of researchers in the pharmacology lab of co-senior author Bryan Roth, M.D., Ph.D., a professor of pharmacology at the University of North Carolina School of Medicine, to identify the most potent of the 23 leading candidates. Then, based on the structural insights of Manglik and Lin, Roth’s team worked with the lab of co-senior author Peter Gmeiner, Ph.D., chair and professor of medicinal chemistry at the Friedrich-Alexander University Erlangen-Nürnberg in Germany, to optimize this compound’s chemical efficacy 1000-fold. This approach succeeded in producing a molecule that the researchers called PZM21, which is chemically unrelated to existing opioid drugs.
‘Unprecedented, weird and cool’ new biology
In further pharmacological tests conducted in the Roth lab, PZM21 exhibited the “new biology” the researchers had been looking for: efficiently blocking pain without producing the constipation and breathing suppression typical of traditional opioids. In addition, PZM21 appeared to dull pain by affecting opioid circuits in the brain only, with little effect the on opioid receptors in the spinal cord that mediate pain reflexes. No other opioid has such a specific effect, Shoichet said, calling it “unprecedented, weird and cool.”
Additional behavioral tests in mice suggested the drug may also lack the addictive qualities of existing opioids. Specifically, the drug didn’t produce the hyperactivity other opioids trigger in mice by activating the brain’s dopamine systems — which are also involved in addiction. Perhaps more tellingly, mice did not spend more time in test chambers where they had previously received doses of PZM21 — a test called “conditioned place preference” that is considered a correlate of human drug-seeking behavior.

Credit: UCSF
“We haven’t shown this is truly non-addictive,” Shoichet cautioned, emphasizing that further experiments in rats and humans would be needed to establish the compound’s addictive potential. “At this point we’ve just shown that mice don’t appear motivated to seek out the drug.”
The study is a successful example of the structure-based approach to drug discovery, a technique partially pioneered at UCSF 30 years ago, Shoichet said, and is one of the first to use structural knowledge to create fundamentally new biological effects.
“This promising drug candidate was identified through an intensively cross-disciplinary, cross-continental combination of computer-based drug screening, medicinal chemistry, intuition and extensive preclinical testing,” Kobilka said.
“If you took away any one of these collaborators it simply wouldn’t have worked,” Shoichet added. “Without Kobilka’s structure, our computation, Roth’s pharmacology, and Gmeiner’s ability to put an atom in exactly the place you want it, this never would have been possible.”
Lead authors on the new paper were Aashish Manglik, M.D., Ph.D., of Stanford University School of Medicine; Henry Lin, Ph.D., of the UCSF School of Pharmacy; and Dipendra K. Aryal, Ph.D., of the UNC School of Medicine. Lin is now principal scientist at The Janssen Pharmaceutical Companies, a division of Johnson & Johnson. Manglik, Lin, Gmeiner, Kobilka, Roth, Shoichet, and co-author Dengler have filed a provisional patent on PZM21 and related molecules, and Manglik, Gmeiner, Kobilka, Roth and Shoichet are consultants and co-founders of Epiodyne, a company seeking to develop novel analgesics.
The research was supported by the National Institutes of Health grants GM106990 (B.K.K., B.K.S. and P.G.), DA036246 (B.K.K.), GM59957 (B.K.S.), and the National Institutes of Mental Health Psychoactive Drug Screening Program (B.L.R.) and DA017204 (B.L.R., D.A.), DA035764 (B.L.R.) and the Michael Hooker Distinguished Professorship (B.L.R.) and the German Research Foundation Grants Gm 13/10 and GRK 1910 (P.G). H.L. received a predoctoral fellowship from the PhRMA Foundation and A.M. received support from the Stanford University Medical Scientist Training Program (T32GM007365) and the American Heart Association (12PRE8120001).
UCSF is a leading university dedicated to promoting health worldwide through advanced biomedical research, graduate-level education in the life sciences and health professions, and excellence in patient care. It includes top-ranked graduate schools of dentistry, medicine, nursing and pharmacy; a graduate division with nationally renowned programs in basic, biomedical, translational and population sciences; and a preeminent biomedical research enterprise. It also includes UCSF Health, which comprises two top-ranked hospitals, UCSF Medical Center and UCSF Benioff Children’s Hospital San Francisco, and other partner and affiliated hospitals and healthcare providers throughout the Bay Area.

